Bienvenue sur le site du GFRS, Le site francophone des spécialistes de la sclérodermie .
Qui sommes nous?

Le Groupe Francophone de Recherche sur la Sclérodermie (GFRS) a été créé en 1996. Il réunit des médecins, paramédicaux et chercheurs motivés par la recherche sur la sclérodermie.
Ses objectifs sont de promouvoir la recherche sur la sclérodermie, qu’elle soit clinique ou translationnelle, d’informer tous les partenaires impliqués, et d’améliorer le soin aux patients atteints de sclérodermie des progrès en cours, en France et dans toute la francophonie.
Le bureau du GFRS est organisé en groupes de travail dont vous pourrez prendre connaissance de la composition et des objectifs sur notre site.
Ce site est le vôtre et est dédié à la sclérodermie systémique dans le monde francophone.
Pr Luc Mouthon, Président du GFRS
Faire un don pour la recherche...(Cliquez pour ouvrir)
Nous vous remercions de l’aide apportée à la recherche contre cette maladie.
Vous pouvez adresser vos dons au Groupe francophone de recherche sur la sclérodermie:
– Par chèque A l’ordre du GFRS
Pr L. MOUTHON,
Centre des maladies rares
Hôpital COCHIN
27 rue du Faubourg Saint-Jacques
Bâtiment Saint-Jacques
75014 Paris
Mail : contact.gfrs@gmail.com
- -Par virement : Agence BNP Paribas Paris Port Royal (00164)
- -En Ligne. en cliquant sur l’icône ‘ faire un don pour la recherche’
 .
.
Nous vous enverrons un justificatif permettant de déduire votre don.
Qu'est ce que la sclérodermie? ...(Cliquez pour ouvrir)
Qu’est ce que c’est que la sclérodermie ?
Les explications du Groupe Francophone de Recherche sur la Sclérodermie (GFRS)
Par les Dr Christian AGARD, Dr Elisabeth Diot, Pr Dominique Farge, Pr Pierre-Yves Hatron, Dr Nathalie Lambert,Pr Luc Mouthon.
La sclérodermie systémique est une maladie rare de cause inconnue. Son nom vient du grec « sclero » qui signifie « dur » et « derme », la peau. C’est donc la maladie de la « peau qui devient dure ». La sclérose atteint la peau et les muqueuses, ainsi que certains organes, notamment les poumons.
- La sclérodermie localisée
Il existe une maladie strictement cutanée, appelée sclérodermie localisée, se présentant généralement sous la forme de plaques d’épaississement de la peau, dénommées « morphées ». Il ne s’agit pas d’une sclérodermie systémique. Les morphées sont des maladies très différentes de la sclérodermie systémique, qui n’entrainent pas d’atteinte des organes profonds.
- La sclérodermie systemique
Au cours de la sclérodermie systémique, tous les organes peuvent être atteints, mais ceci n’est pas constant et certaines atteintes (peau, poumon, tube digestif) sont plus fréquentes que d’autres (cœur, rein) :
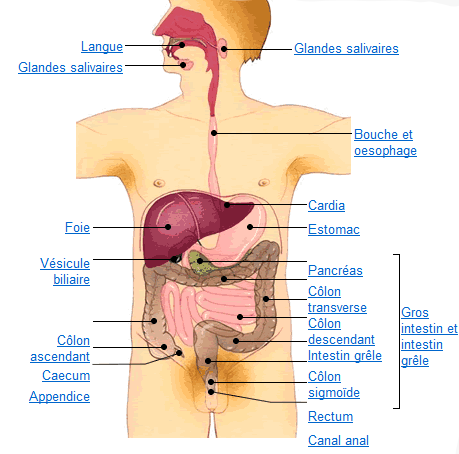
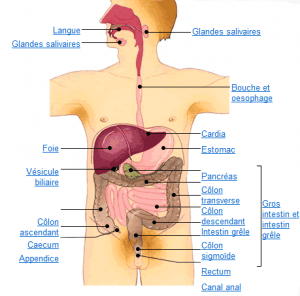 Le tube digestif du haut en bas:
Le tube digestif du haut en bas:
- bouche,
- oesophage,
- estomac, et intestin grêle
- côlon, rectum et anus
- Les autres organes
- Le coeur et les poumons
- Les reins et la vessie
- Les organes génitaux
- Les muscles, nerfs, tendons et articulations
- Le foie
- Le système nerveux central
Les mains et le visage sont des cibles favorites de la sclérodermie systémique. La fatigue est fréquente, les douleurs et les ulcérations des doigts sont sources de handicap au quotidien.
Un bilan complet doit être fait dès que le diagnostic est affirmé et répété chaque année pour suivre l’évolution de la maladie.
L’installation de la maladie peut être longue et insidieuse. Le premier signe est le plus souvent un phénomène de Raynaud, qui précède les autres signes de sclérodermie systémique de 5 ans en moyenne.
Les signes de la sclerodermie:
Les signes amenant au diagnostic sont : l’aggravation du phénomène de Raynaud, un gonflement et un enraidissement des doigts, des ulcérations des doigts, des brûlures au niveau de l’œsophage, ou un essoufflement inhabituel.
Le plus souvent, l’épaississement de la peau (sclérose, ou fibrose) ne concerne que les doigts. Si cet épaississement de la peau ne remonte pas au delà des coudes aux membres supérieurs et des genoux aux membres inférieurs, il s’agit alors d’une sclérodermie systémique cutanée limitée. Lorsque la sclérose s’étend au-delà des coudes et/ou des genoux, ou atteint le tronc, on parle de sclérodermie systémique diffuse.
La sclerodermie, qu’est ce que c’est?
La sclérodermie systémique est une maladie complexe, caractérisée par un excès de fibrose dans certains tissus, des anomalies des petits vaisseaux artériels, principalement des doigts, des perturbations du système immunitaire comme la production d’auto-anticorps.
Ainsi, la sclérodermie systémique est une maladie auto-immune et elle fait partie d’un groupe de maladies appelées « connectivites » (ou collagénoses). Dans le sang des patients, on recherche des anticorps qui sont dirigés contre des protéines présentes dans le noyau des cellules (anticorps anti-nucléaires), qui peuvent être des anticorps anti-centromères, plus volontiers associés aux formes limitées, des anticorps anti-topoisomérase 1 (ou Scl-70), généralement associés à la fibrose pulmonaire et/ou et des anticorps anti-RNA polymérase III, associés aux formes diffuses.
Les mécanismes précis qui contribuent au déclenchement, à l’entretien, ou à la survenue de complications de la maladie ne sont pas tous bien compris mais sont désormais très étudiés par de nombreuses équipes à travers le monde.
La sclérodermie systémique atteint surtout les femmes et concerne 6500 à 10.000 personnes en France. Elle n’est pas contagieuse et ne se transmet pas aux enfants, même si un terrain génétique favorisant des perturbations auto-immunes est très rarement retrouvé au sein de la famille d’un patient. Certaines expositions professionnelles, par exemple à la silice, peuvent favoriser l’apparition d’une sclérodermie systémique.
La fibrose est due à une augmentation de la production de collagène par des cellules, les fibroblastes du derme de la peau ou des autres tissus. Quand la fibrose touche les poumons, on parle de pneumopathie interstitielle, qui est fréquente, et parfois grave. Les petits vaisseaux artériels se contractent et ont une paroi épaissie, ce qui gêne la circulation du sang. Ces anomalies vasculaires sont responsables du phénomène de Raynaud, de certaines ulcérations digitales, de l’atteinte rénale dont la survenue est probablement favorisée par les corticoïdes (crise rénale sclérodermique, rare mais parfois grave), et de l’hypertension artérielle pulmonaire (HTAP, qui concerne 10% des patients et qui peut être grave) quand les petites artères pulmonaires sont touchées.
Traitement
On ne sait pas guérir la sclérodermie systémique, mais il est faux de dire que l’on ne peut rien y faire comme on l’entend parfois. Des possibilités de traitement existent, spécifiques pour chaque symptôme et adaptées à chaque malade. Des progrès importants ont été réalisés ces 10 dernières années dans la prise en charge et le traitement médicamenteux des complications de la sclérodermie systémique que sont les ulcères des doigts, l’HTAP, la pneumopathie interstitielle, la crise rénale. Il n’existe pas de modèle évolutif unique de la maladie, certains patients développant de nombreuses complications, et d’autres n’en développant aucune. Aucune complication n’est inéluctable.
Le GFRS
Le GFRS (Groupe Francophone de Recherche sur la Sclérodermie) est composé de médecins de diverses spécialités (internistes, dermatologues, médecins vasculaires, rhumatologues, pneumologues, pédiatres…) et de chercheurs qui travaillent ensemble pour contribuer à l’amélioration de la connaissance et de la prise en charge des patients atteints de sclérodermie. Le GFRS a toujours eu le soutien de l’ASF (Association des Sclérodermiques de France), ce qui permet d’établir un lien entre la clinique, la recherche et les malades.”
” Le GFRS a la volonté d’établir un lien fort entre les médecins cliniciens, les chercheurs et les patients”
Avertissement: Ce site a pour but de vous expliquer les différentes complications possibles au cours de la sclérodermie systémique. C’est à dire que TOUS LES RISQUES, TOUTES LES COMPLICATIONS sont évoqués et expliqués. Bien sûr, aucun patient atteint de sclérodermie systémique n’a TOUS ces symptômes et le fait d’avoir un organe atteint ne veut pas dire que les complications les plus graves vont survenir.
Pr Christian AGARD, Dr Elisabeth Diot, Pr Dominique Farge, Pr Pierre-Yves Hatron, Dr Nathalie Lambert, Pr Luc Mouthon.
Le bureau du GFRS...(Cliquez pour ouvrir)
Le bureau du GFRS
Le bureau a été élu le 13 Janvier 2015 par les différents membres du Conseil d’Administration.
Pr Luc Mouthon (Hôpital Cochin, Paris), Président
Pr Pierre Yves Hatron (CHRU de Lille), Vice Président,
Dr Nathalie Lambert: ( inserm Marseille) Vice présidente dédiée à la recherche
Dr Elisabeth Diot (CHU de Tours), Secrétaire,
Dr Bernard Imbert (CHU Grenoble), Trésorier
Le nouveau Bureau remercie les électeurs.
Le Conseil d’Administration du gfrs
Le conseil d’administration du GFRS a été élu le 15 Octobre 2014 par les adhérents en Assemblée générale.
- Dr A. Abdessemed – Hôpital de Ben Aknoun
- Pr C. Agard – CHU Hôtel Dieu/ CHU Nantes
- Dr B. Bader-Meunier – Hôpital Necker
- Dr C. Deligny – CHU Fort de France
- Pr D. Farge-Bancel (Hôpital Saint-Louis, Paris)
- Pr B.Granel – Hopitaux Nord de Marseille
- Dr C. Grange – Centre Hospitalier.Lyon Sud
- Dr D. Launay – CHRU Hôpital Cl. Huriez – Lille
- Dr I. Marie – CHU Rouen-Bois guillaume
- Pr T. Martin – CHU Strasbourg
- Dr L. Michel – Labo immunogénétique INSERM – Paris
- Dr A. Solanilla – Bordeaux
Les membres associés:
- SFD: MS Doutre
- SFM: I Lazareth
- SFRF: J Sibilia
- SNFMI: P Roblot
- SPLF: H Numes