Les explications du Groupe Francophone de Recherche sur la Sclérodermie (GFRS) Maj 2024
Par les Professeurs David Launay et Luc Mouthon
Définition et manifestations cliniques
La sclérodermie systémique est une maladie auto-immune rare dont l’origine demeure inconnue. Cette affection atteint principalement les femmes, touchant entre 6 000 et 12 000 personnes en France.
Le terme “sclérodermie” vient du grec sklêros, « dur, sec », et derma, « peau ». La maladie se caractérise par une fibrose avec un épaississement progressif de la peau et potentiellement d’autres organes. Il est important de distinguer la sclérodermie systémique de la sclérodermie localisée, cette dernière étant une affection cutanée stricte, qui se manifeste généralement sous forme de plaques de fibrose cutanée localisées, appelées “morphées”.
Au cours de la sclérodermie systémique, tous les organes peuvent potentiellement être affectés. Cependant, certaines atteintes (comme celle de la peau, du poumon, et du tube digestif) sont plus fréquentes que d’autres (comme celle du cœur et du rein), et certains patients n’ont aucune complication.
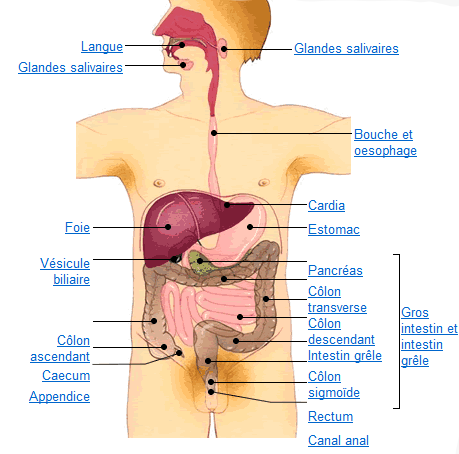
Vous trouverez ci-dessous un récapitulatif des atteintes possibles des différents organes. En cliquant sur chaque organe, vous serez redirigé vers des pages dédiées qui les détaillent.
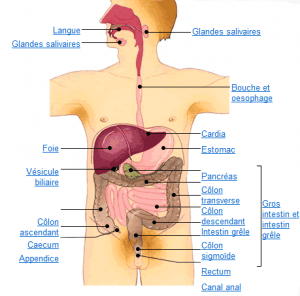 Le tube digestif du haut en bas:
Le tube digestif du haut en bas:
- bouche,
- oesophage,
- estomac, et intestin grêle
- côlon, rectum et anus
- Les autres organes
- Le coeur et les poumons
- Les reins et la vessie
- Les organes génitaux
- Les muscles, nerfs, tendons et articulations
- Début de la maladie
La sclérodermie systémique débute le plus souvent par un phénomène de Raynaud, puis apparaît une atteinte cutanée associée ou non à une ou plusieurs atteintes viscérales. Le délai entre le début du phénomène de Raynaud et les premiers signes de la maladie est variable : il peut être court dans les formes les plus agressives mais il peut excéder plusieurs dizaines d’années dans certains cas. En moyenne, ce délai est de 5 à 10 ans.
Le plus souvent, l’épaississement de la peau ne concerne que les doigts (sclérodactylie). Si cet épaississement de la peau ne remonte pas au-delà des coudes aux membres supérieurs et des genoux aux membres inférieurs, il s’agit alors d’une sclérodermie systémique cutanée limitée. Lorsque la sclérose s’étend au-delà des coudes et/ou des genoux, ou atteint le tronc, on parle de sclérodermie systémique cutanée diffuse. Cette dichotomie clinique garde une pertinence en termes de pronostic et de risque de complications : la sclérodermie systémique cutanée diffuse a un pronostic plus sévère et des atteintes viscérales plus fréquentes.
- Quel est la démarche diagnostique devant une suspicion de sclérodermie systémique ?
Le bilan diagnostique va s’appuyer sur un examen clinique complet, une prise de sang pour rechercher la présence d’auto-anticorps spécifiques et une capillaroscopie (examen du lit de l’ongle au microscope) pour rechercher des mégacapillaires (capillaires géants). Un bilan plus complet doit être ensuite proposé, idéalement au sein des centres de références et des centres de compétence (filière FAI2R), si le diagnostic est posé ou pour conforter ce diagnostic, à la recherche en particulier des complications pulmonaires (pneumopathie interstitielle diffuse et/ou hypertension artérielle pulmonaire). Chez les patients chez qui le diagnostic de sclérodermie systémique est retenu, ce bilan sera répété régulièrement lors du suivi pour dépister une éventuelle complication.
- Quels sont les mécanismes à l’origine de la sclérodermie systémique ?
Il ne s’agit ni d’une maladie contagieuse ni d’une maladie génétique, même si un terrain génétique favorisant des perturbations auto-immunes a été identifié.
Certaines expositions professionnelles (par exemple à la silice ou aux solvants) sont possiblement associées à la sclérodermie systémique, qui peut alors être reconnue dans certains cas comme une maladie professionnelle
La sclérodermie systémique fait partie d’un groupe de maladies appelées « connectivites » (ou collagénoses). C’est une maladie complexe, caractérisée par un excès de fibrose dans certains tissus, des anomalies des petits vaisseaux artériels (vasculopathie), principalement des doigts, des perturbations du système immunitaire comme en témoigne la production d’auto-anticorps. Ces anticorps sont dirigés contre des protéines présentes dans le noyau des cellules (anticorps anti-nucléaires), qui peuvent être des anticorps anti-centromères, plus volontiers associés aux formes limitées, des anticorps anti-topoisomérase 1 (ou Scl-70), généralement associés à la fibrose pulmonaire et/ou des anticorps anti-RNA polymérase III, associés aux formes cutanées diffuses.
Les mécanismes précis qui contribuent au déclenchement, à l’entretien, ou à la survenue de complications de la maladie ne sont pas entièrement compris mais sont étudiés activement par de nombreuses équipes en France et à travers le monde.
La fibrose est due à une augmentation de la production de collagène par des cellules appelées fibroblastes, du derme de la peau ou des autres tissus. Quand la fibrose atteint les poumons, on parle de pneumopathie interstitielle ou fibrose pulmonaire, qui est fréquente, et parfois grave. Les petits vaisseaux artériels se contractent et ont une paroi épaissie, ce qui gêne la circulation du sang. Ces anomalies vasculaires sont responsables du phénomène de Raynaud, de certaines ulcérations (plaies) digitales, de l’atteinte rénale dont la survenue est probablement favorisée par la prise de corticoïdes (crise rénale sclérodermique, rare mais grave), et de l’hypertension artérielle pulmonaire ( 10% des patients) quand les artères pulmonaires sont concernées.
- Traitement
On ne sait actuellement pas guérir la sclérodermie systémique, mais il est faux de dire que l’on ne peut rien y faire et qu’il n’y a pas de traitement comme on l’entend encore parfois. Des possibilités de traitement existent, spécifiques pour chaque symptôme et adaptées à chaque malade. Des progrès importants ont été réalisés ces 10 dernières années dans la prise en charge et le traitement médicamenteux des complications de la sclérodermie systémique que sont les ulcères digitaux, l’hypertension artérielle pulmonaire, la pneumopathie interstitielle, ou la crise rénale. La rééducation et la pratique régulière d’exercices et d’activités physiques permettent d’améliorer la qualité de vie. Des progrès sont également faits dans la prise en charge globale de la maladie, que ce soit sur les choix et les options thérapeutiques ou sur l’éducation thérapeutique du patient.
Récemment, un protocole national de diagnostic et de soins a été publié et on peut le retrouver sur le site de la filière FAI2R.
Le GFRS
Le GFRS (Groupe Francophone de Recherche sur la Sclérodermie) est composé de médecins de diverses spécialités (internistes, dermatologues, médecins vasculaires, rhumatologues, pneumologues, pédiatres…) et de chercheurs qui travaillent ensemble pour contribuer à l’amélioration de la connaissance et de la prise en charge des patients atteints de sclérodermie systémique. Le GFRS a toujours eu le soutien de l’ASF (Association des Sclérodermiques de France) et réciproquement, ce qui permet maintenir le lien entre le soin, la recherche et les patients.
Avertissement: Ce site a pour but de présenter la sclérodermie systémique et ses complications possibles. Si de nombreuses complications sont évoquées, un patient avec une sclérodermie systémique peut n’en présenter aucune et le fait d’avoir un organe atteint ne veut pas nécessairement dire que les complications les plus graves vont survenir.