Fairchild R, Chung M, Yang D, Sharpless L, Li S, Chung L.
Development and Assessment of a Novel Lung Ultrasound Interpretation Criteria for the Detection of Interstitial Lung Disease in Systemic Sclerosis. Arthritis Care Res [Internet]. 31 mai 2020 [cité 15 juin 2020]; (PubMed)

Rédacteur : Dr Sébastien SANGES (Lille)
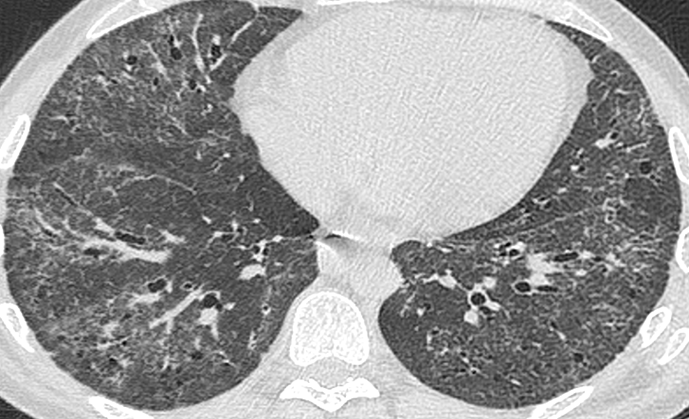
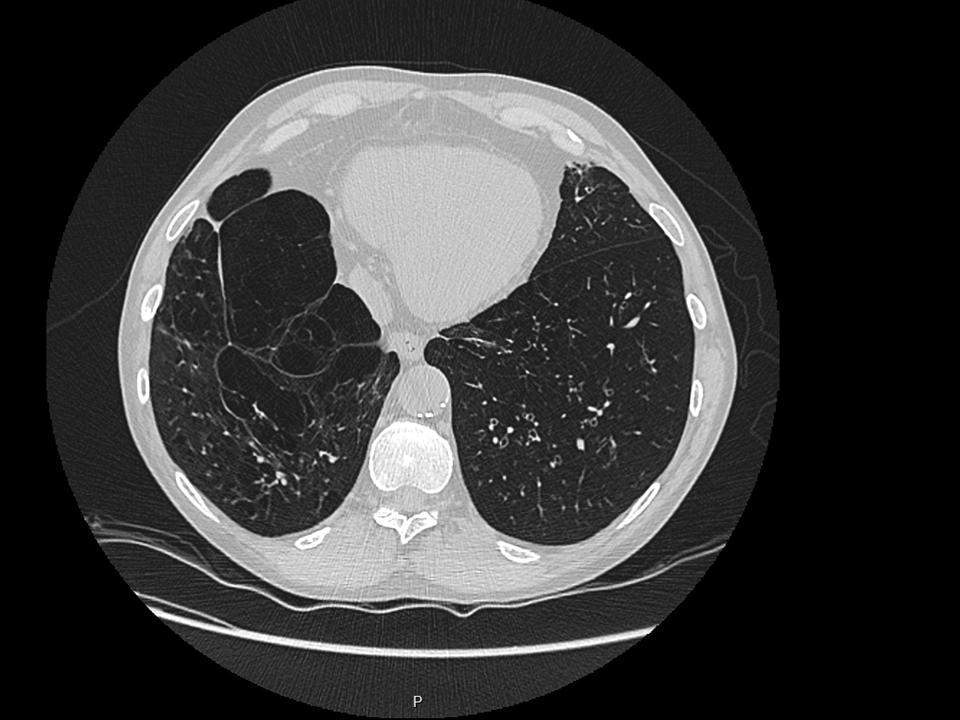
La pneumopathie interstitielle diffuse (PID) est une complication fréquente et potentiellement grave de la sclérodermie systémique (SSc). Son dépistage passe par la réalisation du scanner thoracique, examen irradiant, et d’épreuves fonctionnelles respiratoires, qui manquent de sensibilité pour les formes débutantes.
Dans cette étude prospective américaine, les auteurs ont étudié l’intérêt du dépistage de la PIDSSc par échographie pulmonaire, en le comparant à la méthode gold-standard du scanner thoracique.
Au total, 20 patients SSc (9 formes diffuses, 11 formes limitées) étaient inclus. Tous ont bénéficié d’un scanner thoracique et d’une échographie pulmonaire selon un protocole standardisé (acquisition de boucles de 4 secondes sur 14 positions prédéfinies par 2 évaluateurs en aveugle ; interprétation selon des critères standardisés).
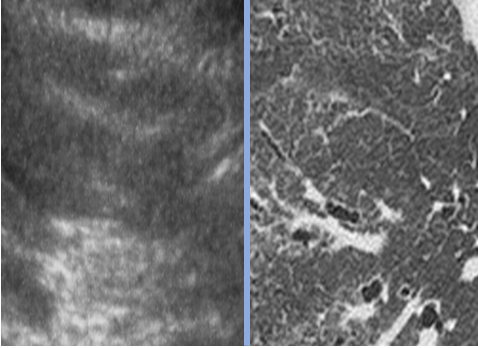
Le scanner thoracique mettait en évidence une PID chez 9 patients (45%, 8 PINS et 1 PIC).
L’échographie pulmonaire retrouvait des signes de PID chez 11 patients (55%), soit une sensibilité de 100% et une spécificité de 82% par rapport au scanner. La concordance entre les 2 échographistes était complète (100%) pour le diagnostic de PID et excellente (94%) pour l’interprétation des 278 images acquises.
Les auteurs suggèrent ainsi que leur protocole standardisé d’échographie pulmonaire constitue une méthode très sensible et spécifique de dépistage de la PID-SSc, avec une bonne homogénéité entre les examinateurs.