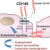
L’HTAP est une maladie chronique caractérisée par une élévation progressive de la pression au niveau de la circulation artérielle pulmonaire, c’est à dire les vaisseaux sanguins qui partent du coeur droit (ventricule droit) pour amener du sang veineux, pauvre en oxygène, vers les poumons. Dans l’HTAP, la résistance à l’écoulement de ce sang est anormalement augmentée et l’évolution se fait vers une diminution progressive du débit cardiaque et une insuffisance cardiaque droite, qui peut être fatale en l’absence de traitement.
L’HTAP est une maladie chronique caractérisée par une élévation progressive de la pression au niveau de la circulation artérielle pulmonaire, c’est à dire les vaisseaux sanguins qui partent du coeur droit (ventricule droit) pour amener du sang veineux, pauvre en oxygène, vers les poumons. Dans l’HTAP, la résistance à l’écoulement de ce sang est anormalement augmentée et l’évolution se fait vers une diminution progressive du débit cardiaque et une insuffisance cardiaque droite, qui peut être fatale en l’absence de traitement.
L’HTAP est la conséquence d’anomalies des petites artères pulmonaires qui se contractent de manière excessive et irréversible (excès de contraction lié à des molécules comme l’endothéline 1, défaut de dilatation par manque de molécules comme le NO ou la prostacycline), et qui sont également anormalement épaissies (remodelage vasculaire), ce qui favorise l’obstruction progressive de ces vaisseaux sanguins.
Pour affirmer puis suivre l’évolution d’une HTAP, il est nécessaire de faire une mesure précise des pressions artérielles pulmonaires (PAP) en introduisant une sonde dans les artères pulmonaires lors d’un examen appelé « cathétérisme cardiaque droit ». Dans l’HTAP la PAP moyenne (PAPm) est supérieure à 25 mm Hg (normale<20 mmHg).
L’HTAP est une maladie existant en tant que tel (HTAP dite « idiopathique »), mais elle représente aussi une complication possible de certaines maladies auto‐immunes, principalement la sclérodermie systémique (ScS). L’HTAP est une maladie rare et la fréquence de l’HTAP au cours de la ScS est évaluée à 8‐15 %. Elle survient le plus souvent dans les 5 à 10 années qui suivent le diagnostic de ScS, mais peut s’observer au diagnostic initial de ScS ou de nombreuses années après. L’HTAP concerne un peu plus souvent les formes limitées de ScS, mais les formes diffuses peuvent également se compliquer d’HTAP.
Une HTAP doit être évoquée chez tout patient sclérodermique quand il existe un essouflement anormal, une fatigue intense, des palpitations, des oedèmes des membres inférieurs ou des malaises. Du fait de sa gravité, l’HTAP doit en fait être dépistée chez tout patient sclérodermique, même en l’absence de ces symptômes. Le dépistage repose sur la réalisation annuelle d’une échographie cardiaque. Si cet examen suspecte une HTAP, un cathétérisme cardiaque droit doit alors être réalisé
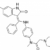
La prise en charge thérapeutique de l’HTAP est complexe et il existe des traitements spécifiques de l’HTAP, qui favorisent la dilatation des petites artères pulmonaires et qui tentent de bloquer les lésions de remodelage de ces vaisseaux. Ces traitements sont ciblés sur les trois principales voies métaboliques impliquées dans l’HTAP et 3 familles de traitement sont donc disponibles : les inhibiteurs des récepteurs de l’endothéline 1 (bosentan, ambrisentan), les inhibiteurs de la phosphodiestérase 5 qui jouent sur la voie du NO (sildénafil, tadalafil) et les analogues de la prostacycline (époprosténol, iloprost, tréprostinil). A la différence des autres, les analogues de la prostacycline ne sont pas disponibles par voie orale mais nécessitent une pompe pour une administration en continu (époprosténol IV, tréprostinil SC) ou de fréquents aérosols (iloprost). Grâce à l’ensemble de ces médicaments, le pronostic de l’HTAP de la ScS s’est amélioré depuis 10 ans environ, mais ces traitements sont encore insuffisamment efficaces et des progrès sont espérés. Chez certains malades, une transplantation pulmonaire ou cardio‐pulmonaire peut parfois être envisagée.