Khanna D et al, American Journal of Respiratory and Critical Care Medicine 2022 PMID: 34851799 DOI: 10.1164/rccm.202103-0714OC
Analyse par Magali Lebrun sous la direction de Brigitte Granel
Contexte
La sclérodermie systémique (ScS) est une maladie auto-immune rare caractérisée par une fibrose diffuse et des anomalies vasculaires affectant notamment la peau et des poumons. Le taux moyen de mortalité à 10 ans est de 34%, soit le plus élevé parmi les pathologies rhumatologiques, en lien avec la pneumopathie interstitielle diffuse (PID).
Des taux élevés d’Interleukine 6 (IL-6), une cytokine pro-inflammatoire, sont retrouvés chez les patients avec une forme cutanée diffuse ou une PID de la ScS, et sont associés au déclin de la Capacité Vitale Forcée (CVF). Ainsi l’IL-6 serait un facteur clé de la progression de la maladie au stade précoce.
Le Tocilizumab est un anticorps monoclonal anti récepteur de l’IL-6 : dans l’essai de phase 2 faSScinate (tocilizumab SC pendant 24 semaines), l’efficacité sur l’amélioration de la fibrose cutanée n’a pas été atteinte mais le groupe tocilizumab bénéficiait d’un ralentissement du déclin de la CVF. Dans l’essai de phase 3 focuSSced, le Tocilizumab n’a pas non plus montré d’amélioration significative du score cutané à 48 semaines (S48), mais une amélioration de la CVF et de la fibrose pulmonaire sur la TDM. Les auteurs rapportent l’efficacité et la sécurité à long terme (S96) du Tocilizumab suite à la période supplémentaire de 48 semaines en ouvert de l’essai focuSSced.
Objectif
Les auteurs ont analysé l’efficacité et la sécurité du Tocilizumab à S96 selon le score cutané de fibrose, la variation de CVF et de DLCO, l’évaluation globale de la qualité de vie ainsi que le nombre d’événements indésirables (EI) et d’EI graves pour 100 patients année.
Matériel et méthodes
Il s’agit d’une étude multicentrique, randomisée, contrôlée, en double aveugle (jusque S48) puis en ouvert (jusque S96) portant sur des patients adultes en Europe, en Amérique et au Japon.
Les patients inclus répondaient aux critères de l’ACR-EULAR 2013 et avaient une atteinte cutanée diffuse, un début de la ScS de moins de 5 ans et un syndrome inflammatoire biologique ; entre 61 et 65 % avaient une PID préexistante.
L’étude évaluait l’efficacité et la sécurité à S96 du Tocilizumab par rapport à un placebo, sur la progression des atteintes cutanée (critère principal) et pulmonaire (critère secondaire).
Résultats
Parmi les 212 patient inclus dans la phase en double aveugle, 107 ont reçu le placebo et 105 le Tocilizumab. Lors de la phase en ouvert, respectivement 89 et 92 patients de chaque groupe (Placebo puis Tocilizumab = PT, et Tocilizumab continu = TC) ont reçu du Tocilizumab pendant 48 semaines supplémentaires. L’âge moyen et la durée de la maladie étaient de 48 ans et 23 mois.
Les patients des 2 groupes présentaient une amélioration comparable du score cutané lors de la phase en ouvert.
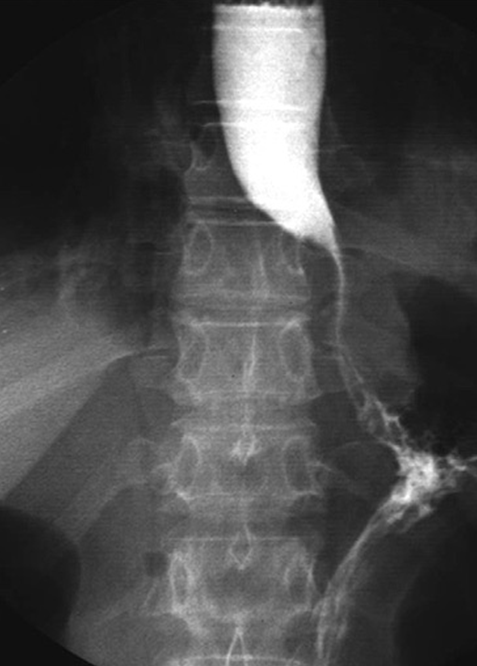
En revanche, il existait une moindre aggravation de la CVF à S96 chez les patients du groupe TC, notamment ceux avec PID préexistante, par rapport au groupe PT (dans la continuité des résultats à S48). Par ailleurs, le déclin de la CVF observé à S48 chez les patients du groupe PT était freiné à S96 après la prise de Tocilizumab.
On ne notait pas de différence de qualité de vie à S96 entre les 2 groupes.
Les taux d’EI et EI graves durant la période en ouvert étaient similaires entre les 2 groupes, stables par rapport à la période en aveugle (taux pour 100 années-patients d’EI graves de S48 à S96 de 14,8 pour PT et de 15,8 pour TC).
Le principal EI relié au Tocilizumab était les infections. Six décès survenaient pendant l’essai dont 2 pendant la période en ouvert, reliés au traitement.
Conclusion
Chez les patients atteints de SSc précoce et de forme cutanée diffuse, le Tocilizumab n’a pas permis d’améliorer l’atteinte cutanée. En revanche, il a permis de préserver la fonction respiratoire à S48 et S96, y compris chez les patients atteints de PID quelle qu’en soit la sévérité. Il existe également une stabilisation du déclin de la CVF sous Tocilizumab à S96 chez les patients ayant initialement présenté une aggravation à S48 sous placebo.
Cette étude montre donc une potentielle efficacité du Tocilizumab sur l’atteinte pulmonaire de la ScS; il est autorisé dans cette indication aux USA, alors qu’en France, il ne peut être envisagé qu’après validation en RCP (utilisation hors AMM).