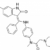
le GFRS soutient les essais cliniques / appels à observation suivants :
– SCLERITA : Essai de phase II évaluant un inhibiteur de JAK, l’itacitinib dans la Sclérodermie Systémique.
Contact : Dr. Benjamin Chaigne benjamin.chaigne@aphp.fr
Appels à Observation :
– SCLEROTRANSPLANT : Transplantation cardiaque en traitement de l’atteinte cardiaque primitive de la sclérodermie systémique en France
Contacts: Dr. Arthur Renaud arthur.renaud49@gmail.com et Pr. Christian Agard christian.agard@chu-nantes.fr
– SCLEROJAKI : Etude non interventionnelle sur données pré-existantes pour évaluer l’efficacité des inhibiteurs de JAK au cours des PID associées à la sclérodermie systémique
Contact: Dr. Paul Decker p.decker@chru-nancy.fr
– SCLEROVID: Exacerbation aiguë de fibrose pulmonaire contemporaine / dans les suites d’une infection à SARS-CoV-2 au cours de la sclérodermie systémique: étude rétrospective française
Contact: Dr. Benjamin Thoreau benjamin.thoreau@aphp.fr
– FEOTOX : État des lieux de la prise en charge des patients développant / aggravant une fasciite avec éosinophiles sous immunothérapie anti-cancéreuse
Contact : Dr. Benjamin Chaigne benjamin.chaigne@aphp.fr
– SCLEROTOX : État des lieux de la prise en charge des patients développant une sclérodermie systémique de novo sous immunothérapie anti-cancéreuse
Contact : Dr. Benjamin Chaigne benjamin.chaigne@aphp.fr
– SCLEROANTIFIBROTIC: Etude nationale multicentrique sur l’utilisation des antifibrosants en vie réelle dans la pneumopathie interstitielle diffuse liée à la sclérodermie systémique
Contacts: Pr. David Launay, Pr. Yurdagul Uzunhan, Dr Vincent Koether, david.launay@chru-lille.fr;yurdagul.uzunhan@aphp.fr; vincent.koether@univ-lille.fr
– MYCRISS : Evaluation de l’association rituximab-mycophenolate mofetil dans la sclérodermie systémique : une étude rétrospective Française multicentrique
Contacts: Dr François Barde et Dr. Benjamin Chaigne : francois.barde@aphp.fr; benjamin.chaigne@aphp.fr
N’hésitez pas à contacter les investigateurs pour toute question et/ou discuter/partager sur ces essais cliniques et appels à observation.